11380 1
Несовершенный остеогенез (osteogenesis imperfecta) — врожденная ломкость костей. Это сложное заболевание костей и некоторых соединительнотканных структур, имеющее широчайший диапазон изменений, известно с глубокой древности как заболевание с выраженной клинической картиной и различными формами, передающееся по наследству. Первые упоминания о нем появились в XVII в. В конце XVIII в., т.е. 200 лет назад, Olaus Jacob Ekmann описал НО у членов одной семьи, N. Ekroth (1788) сообщил о заболевании, которое в четырех семьях передавалось детям, и назвал его osteomalacia congenita. Axmann (1831) не только описал ломкость костей у себя и брата, но и, очевидно, первый отметил такой важный симптом, как наличие голубых склер.
Lobstein (1833) описал ломкость костей у больных различного возраста. По данным Vrolik (1849), переломы у детей происходили или еще внутриутробно, или вскоре после рождения. Е. Looser (1906) описал эти две формы как osteogenesis imperfecta congenita und tarda.
Изучением заболевания занимались многие врачи, описавшие более 20 различных симптомов, из которых основными являются:
изменения в строении скелета и легко наступающие переломы, часто небольшой рост; голубые склеры; опаловидный дентин (dentinogenesis imperfecta); прогрессирующая деформация позвоночника, грудной клетки, черепа и длинных трубчатых костей; тугоухость по проводниковому типу; гиперэкстензия в суставах и их деформация; изменения со стороны сердца и крупных сосудов, носовые кровотечения и др.
Работами последних лет показано, что несовершенный остеогенез является гетерогенным наследственным заболеванием генетической природы, поражающим соединительную ткань и выражающимся остеопенией и вышеперечисленными клиническими признаками.
Вместо двух форм, или типов, в настоящее время по предложенной в 1979 г. D.O. Sillence классификации несовершенный остеогенез с учетом клинических, рентгенологических и коллагеновых протеин-генных молекулярных изменений подразделяют на 4 типа.
Тип I — слабовыраженная форма, доминантно-наследственный несовершенный остеогенез с ломкостью костей и голубыми склерами.
Тип II — перинатально-летальный.
Тип III — прогрессирующее деформирование скелета.
Тип IV — доминантный с нормальными склерами и нерезко выраженными деформациями.
P.A. Dawson и др. (1999) выявили мутации типа I коллагеновых генов как причину всех четырех типов несовершенного остеогенеза (OI). У 2 детей рентгенограммы показали сниженную костную плотность поясничного отдела позвоночника и множественные переломы по всему позвоночнику; эта патология обусловлена изменениями в белках, особенно коллагена типа I. Энзимные изменения касалась единственной базальной мутации (1715 GA) у этих детей. Такая мутация предсказывает замену аргинина на глицин в позиции п43б (С4збК) в а2 (I), отец ребенка имел мутацию ДНК гена. Существование такой же гетерозиготной мутации у 2 детей предполагает, что пробанды отражают полностью этот фенотип. Клинические, биохимические и молекулярные находки расширяют представления о фенотипе, сочетанном с мутациями типа I коллагена, вызывающими изменения в позвоночнике, карликовость в подростковом возрасте.
На основании литературных публикаций последних лет, а также данных, приведенных на 3-й Международной конференции по несовершенному остеогенезу в 1985 г., и работ D.O. Sillence (1985) и др. приводим краткую характеристику этих 4 типов.
Тип I . Остеопороз и переломы костей чаще наблюдаются в раннем возрасте; после 10 лет частота их возникновения уменьшается и опять увеличивается после 40 лет. Переломы приводят к деформации костей. У 50 % больных отмечается небольшой рост. Голубизна склер усугубляется преждевременным появлением старческого ободка. У части больных дентин не изменен, тогда как у другой части его называют опаловым. Встречаются изменения аорты и митральный порок сердца, носовые кровотечения. У 20 % больных с НО I типа наблюдается пролапс митрального клапана. Такой больной описан И.А. Шамовым и Ш.М. Захарьевским в 1989 г. Эта форма обусловлена структурными мутациями в спиральном домене про-а, возможность передачи по наследству около 7 %.
Тип II. Перинатально-летальный несовершенный остеогенез. Клинически и биохимически это гетерогенная группа больных, для которых характерны внутриутробная или ранняя неонатальная смерть, множественность и легкость наступления переломов. Подразделяется на три группы.
Группа А. Хрупкость соединительнотканных образований настолько выражена, что повреждения конечностей и головы плода происходят еще во время беременности; мозговой череп непропорционально велик, грудная клетка маленькая, конечности укорочены и искривлены, встречаются очень тяжелые степени обызвествления стенок аорты и эндокарда, очень малый рост при рождении (иногда 30—25 см).
Часто преждевременные роды: в 15 % случаев в ягодичном предлежа нии, до 20 % — мертворожденные, остальные умирают или в первые дни, или на 4-й неделе жизни. Рентгенологические изменения определяются у плода еще до рождения: широкие бедренные кости с волнистыми краями, короткая грудная клетка, ребра с четками и т.д. Согласно генетическим данным большинство таких случаев являются спорадическими. Биохимические данные позволяют предположить, что больные группы А «... являются гетерогенными для мутаций, вызывающих нарушение npo-oci (I) коллагеновых цепей, приводя к дефектной triple helical assembly секвестрации и инкорпорации внутрь нормальной соединительной ткани. Малое количество больных обладает гетерозиготными мутациями в npo-ai (I) коллагеновой цепи, в то время как некоторые другие описывались с единичной аминокислотной заменой, т.е. глицина на цистин, приводя к формированию дисульфатных мостиков между двумя цепями cti (I) и избыточному скоплению I типа коллагеновых молекул» . Обследование пробандов говорит о возможном молекулярном дефекте, который совместим с гетерозиготностью мутаций в коллагеновом гене, что проявляется в особенностях наследования — аутосомно-доминантном.
Группа Б по фенотипу похожа на группу А, однако нарушения дыхательной системы менее выражены и больные живут несколько лет. Трубчатые кости укорочены и расширены, изменены ребра, но переломы их редки. Предполагается аутосомно-рецессивная наследственность вследствие свежей мутации.
Группа В наблюдается редко, часто отмечаются мертворожденность и смертность в течение первого месяца жизни. Больные маленького роста, трубчатые кости тонкие, особенно диафизы, отсутствует оссификация в костях мозгового и лицевого черепа. Предполагается аутосомно-рецессивная наследственность.
Тип III встречается относительно редко, тело новорожденных укорочено, масса тела может быть нормальной, переломы иногда происходят в процессе родов, а иногда в возрасте нескольких лет. Формируются деформации конечностей (О-образные), кифосколиоз, особенно прогрессирующие во время пубертатности. Изменения скелета и сердечно-сосудистой системы приводят к смерти 40—50 % больных. Резко выражен остеопороз — остеопения, нарушены оссификация и рост костей в длину, в ростковых зонах костей — неравномерное обызвествление, приводящее к образованию пятнистости («кукурузные зерна»).
Как указывает D.O. Sillence (1985), для этого типа характерна аутосомно-рецессивная наследственность. Только у одного больного он мог констатировать, что фенотип образовался благодаря гомозиготности для молекулярного дефекта в коллагене. Наследственность свежая аутосомная, доминантная мутация или аутосомно-рецессивная.
Тип IV. Изменения в скелете встречаются наиболее часто. Характерна большая вариабельность остеопении, возраста, количества переломов костей, голубизны склер (у взрослых склеры могут быть нормального цвета). Количество переломов с возрастом уменьшается, происходит нормальное образование костной мозоли, в возрасте старше 30 лет у V3 больных нарушается слух. Больные этого типа несовершенного остеогенеза подразделяются на две группы: с резко измененными опаловыми зубами и без изменений зубов. Преобладание аутосомно-доминантной наследственности выражается резко благодаря отсутствию фенотипического маркера (как голубые склеры).
В настоящее время считается, что несовершенный остеогенез обусловлен качественными и количественными изменениями в синтезе коллагена I типа. При I типе несовершенного остеогенеза синтез структурно нормального коллагена снижен, тогда как при II и IV типах синтез такого коллагена бывает нормальным, но из-за пониженной стабильности общее количество коллагена снижается. По данным D.O. Sillence (1985), число коллагеновых молекул, продуцируемых при несовершенном остеогенезе, быстро и постоянно увеличивается, но все же не достигает нормы. Поэтому он считает, что в данном случае наблюдается не простое нарушение синтеза коллагена вследствие изменения 4-й хромосомы, а нарушение свойств соединительной ткани, вызванное изменением и протеогликанового синтеза, и генаколлагена.
D.H. Colin и Р.Н. Byers (1991) обнаружили следующее: у 4 больных из 60 клеток синтезировали популяцию цепи а2 (I) с остатками цистина в тройной спирали, а клинические различия и гетерогенность в локализации остатков цистина дают основание предположить, что положение и места замены внутри самой цепи являются важными в определении клинического фенотипа. Этим подтверждается мнение о том, что больные с нелетальной формой несовершенного остеогенеза могут часто иметь дефекты в COL A1 или в COL 1A2 генах, предполагая, что многие из таких дефектов замещаются на остатки глицина в оа (I) тройного спирального пространства.
L. Cohen-Solal и др. (1991) показали, что тип II и тип III несовершенного остеогенеза могут появиться вследствие гонадного мозаицизма. что очень важно для генетической консультации при определении соответствующего фенотипа заболевания.
Анализы на проколлаген типа I молекул, синтезированных дермальными фибробластами, культивированными от больных с несовершенным остеогенезом, позволили установить две обширные биохимические группы: 1) больные, фибробласты которых синтезировали и эффективно секретировали около половины ожидаемого количества структурно нормального проколлагена I типа ; 2) больные, фибробласты которых продуцировали нормальные и ненормальные популяции молекул и затем секретировали их .
R.J. Wenstrup и др. (1990) сообщили, что они провели аналогичные исследования у 224 больных и сравнили полученные биохимические данные с клинической картиной. Оказалось, что в 1-й группе, где наблюдалось уменьшение количества нормального проколлагена типа I, клинические проявления были небольшими, а во 2-й группе, где обнаруживался синтез нормальных молекул и ненормального проколлагена типа I, фенотип варьировал от умеренно деформирующего кости и со слегка укороченной фигурой до заболевания, резко деформирующего скелет с умеренно или резко укороченной фигурой. Эти и другие исследования позволяют ставить пренатальный диагноза. По мнению R.J. Wenstup и др. (1990), при лечении нужно учитывать биохимические дефекты.
Л.М. Михайлова (1971) при ультрамикроскопическом исследовании костной ткани больных с несовершенным остеогенезом во многих остеобластах отметила редукцию элементов гранулярного эндоплазматического ретикулума, что вызывало нарушение фибриллогенеза; оказывались измененными также митохондрии, в матриксе которых имелись скопления кристаллов (очевидно, гидроксиапатита), что, по ее мнению, свидетельствовало о нарушении кальциевых и фосфатных ионов. По данным М.В. Волкова и Н.Н. Нефедьевой (1974), у больных резко увеличено содержание гексоз, гликопротеидов, гексозаминов, сиалопротеидов в сыворотке крови и с мочой выделяется повышенное количество мукополисахаридов. Патологические изменения у больных с несовершенным остеогенезом весьма разнообразны.
Псевдосаркомы . После перелома развивается костная мозоль больших пли громадных размеров (рис. 5.1), резко поротичная, постепенно, в течение ряда лет или десятилетий, увеличивающаяся, которую приходится дифференцировать от саркомы, тем более что в литературе имеются указания на развитие остеогенной саркомы у больных с НО. Развитие псевдосаркомы сопровождается довольно сильными болевыми ощущениями, напряжением тканей, местной гиперемией.
Развитие костной мозоли больших размеров, по мнению Т.П. Виноградовой (1973), является механизмом, компенсирующим недостаточную прочность ее структур. После срастания отломков эти опухолеподобные мозоли исчезают. Однако очень редко у больных с НО костные мозоли не рассасываются, а остаются необычно большими (какими были первоначально) или медленно продолжают расти, так что их уже невозможно принимать за проявление компенсаторного процесса. Удовлетворительных гипотез их происхождения нет. Мы наблюдали 3 больных с развитием «псевдосарком», у 2 из которых они достигали гигантских размеров.
Рис. 5.1. Костная мозоль, обусловившая увеличение правой бедренной кости, — псевдосаркома.
Одну больную мы оперировали. Костная ткань имела вид спонгиозы с тонкими перегородками и большими лакунами жирового костного мозга.
Создалось впечатление, что разрастание костного мозга приводит к увеличению объема кости, костных лакун, а реактивное костеобразование способно только к образованию тонких перегородок и полостей, но не способно остановить процесс, в связи с чем не может образоваться нормальный кортикальный слой.
Мы считаем допустимым предположить, что при НО наблюдаемая остеопения является следствием, во-первых, некоторого уменьшения количества «активных ячеек роста костной ткани», которые, согласно теории, разработанной Н.М. Frost и др., определяют моделирование костной ткани; во-вторых, следствием изменений в коллагеновых структурах и, в-третьих, очевидно, следствием нарушений обмена в «третьей разновидности жировой ткани». По А.А. Заварзину (1985), такой разновидностью является жировая ткань костного мозга, жировые клетки которого содержат особые липиды, обычно не используемые в липидном обмене. Бурная пролиферация соединительной ткани, наблюдаемая при переломах и развитии псевдосаркомы , способствует образованию больших лакун и тем самым спонгизации кости: на участках, где развивается псевдосаркома, иногда кортикальный слой как таковой не определяется.
А.Н. Черняев и Г.А. Грибанов (1982) показали, что продолжительное введение кальцитонина способствует увеличению синтеза фибробластами не только коллагена, гликозаминогликанов, но и липидов. Естественно, необходимо тщательно исследовать в динамике уровень выработки кальцитонина у больных с псевдосаркоматозными формами несовершенного остеогенеза. Нам пришлось в течение 30 лет наблюдать больную с резко выраженной формой псевдосаркоматозной формы несовершенного остеогенеза. Он протекает не равномерно, а стадийно, период медленного спокойного течения сменяется периодом бурного развития, появляются боли в той или иной кости, местно повышается температура, что сопровождается появлением участков гиперемии без четких границ, резко возрастает уровень щелочной фосфатазы.
Больная А. наблюдалась нами с возраста 33 лет до 61 года. Родилась нормальным ребенком в 1933 г., самостоятельно ходила до 1 года 9 мес, когда произошел перелом правого бедра. Через год — повторный перелом правого бедра, в возрасте 6 лет — перелом костей правой голени, затем левой бедренной кости, всего было 7 переломов. Больную консультировали известные специалисты: Г.С. Бом, П.А. Герцен (сказал — «проживет не больше года»), С.М. Спасокукоцкий, Т.П. Краснобаев («у этой болезни нет названия»), И.Г. Лагунова, М.К. Климова. В 1970 г. обратилась в ЦИТО и была стационирована с диагнозом: несовершенный остеогенез, псевдосаркоматозная форма.
Больная очень маленького роста (107 см), с трудом ходит на костылях, предпочитает передвигаться на каталке. Жалобы на постоянно увеличивающееся в объеме правое бедро, представлявшее собой несколько вытянутый «арбуз», вверху переходивший в таз, а внизу заканчивавшийся у колена. Были увеличены в объеме также большеберцовая кость и левое бедро. Движений в правом тазобедренном суставе практически не было, и больная не могла произвести туалет промежности, а при мочеиспускании моча попадала на внутреннюю поверхность бедра. Нами произведена подвертельная остеотомия правой бедренной кости, при этом понадобился не молоток, а долото, которое под нажимом руки легко погружалось в кость, представлявшую жировой костный мозг, разделенный тонкими костными перегородками. Произведена остеотомия 3 /4 поперечника бедренной кости, после чего нога отведена кнаружи и фиксирована гипсовой лонгетой. Клинически патологическая измененная кость производила впечатление разрастающегося костномозгового жира и остеопорозной истонченной костной ткани: редкие атрофичные костные трабекулы.
В течение 25 лет значительных перемен в состоянии больной не было. В 1995 г. произошел перелом бедренной кости, после чего ее объем стал быстро увеличиваться, как и объем левой голени, больная с трудом переворачивалась на кровати. При осмотре в 1997 г. оба бедра и голени резко увеличены в объеме. Увеличены и все кости таза с обеих сторон, состояние больной тяжелое. Через месяц по телефону мне сообщили, что у нее произошел перелом нескольких ребер, собираются положить в больницу. Связь прервалась.
Лечение . В настоящее время принято считать, что при всех формах НО показано лечение остеопороза витамином D3, комплексонами (ксидифоном и др.), бифосфонатами, глюконатом кальция, глицерофосфатом, солями магния, калия. Реже применялось лечение рыбьим жиром, витамином D2, анаболическими гормонами, ультрафиолетовым облучением [Волков М.Б., Нефедьева Н.Н., 1974]. Большее распространение и эффект давало лечение, разработанное в 1984 г. Н.А. Беловой в виде схемы и рассчитанное на 12 мес (соматотропный гормон по 4 ЕД 3 раза в неделю в течение 1-го и 9-го месяцев; кальцитрин по 3—7,5 ЕД ежедневно в течение 2-го и 10-го месяцев; витамин D2 — 9-й и 12-й месяцы; оксидевит (витамин D3) по 1 — 1,5 мкг в сутки — 3-й, 4-й и 11-й, 12-й месяцы; фестал, панзинорм, глюконат кальция, фитин, цитратная смесь, витамины А, Е, электрофорез с солями кальция, массаж, ЛФК). По данным А.П. Бережного с соавт. (1988), это консервативное лечение позволило получить положительные результаты: у ряда больных прекратились переломы длинных трубчатых костей, а проведенное в предоперационном периоде лечение позволило улучшить результаты операций. Таким образом, консервативное лечение с применением витамина D3 и других препаратов следует проводить всем больным с НО.
Консервативное лечение переломов костей у этой группы больных является довольно сложной задачей, поскольку у некоторых из них переломы возникают часто, а иногда бывают множественными. Необходимо использовать все имеющиеся методы лечения, а иногда ставить показания к оперативному вмешательству.
Учитывая повышенную ломкость костей, некоторые ортопеды для исправления деформации осуществляли остеоклазию на вершине искривления, исправляли деформацию и фиксировали конечность гипсовой повязкой или вытяжением.
Оперативное лечение в 40—50-х годах осуществлялось у единичных больных. Ф.Р. Богданов (1945) производил сегментарные остеотомии, а для интрамедуллярной фиксации применял предложенный им штифт. Т.С. Зацепин использовал штифты из гетерокости и металла. В 1964 г. М.В. Волков предложил в качестве интрамедуллярного фиксатора аллогенные трансплантаты, а затем разработал методику, которая включает декортикацию деформированной кости, сегментарную остеотомию и пластику с помощью аллотрансплантатов по типу «вязанки хвороста». Эта методика оказалась очень эффективной, аллогенные трансплантаты при этом спаиваются остеогенной тканью и постепенно перестраиваются.
В руководимом нами отделении оперативное лечение произведено 43 таким больных, которым в общей сложности выполнено 91 оперативное вмешательство. Ортопедам, занимающимся оперативным лечением больных с НО, приходится учитывать изменения скелета у больного и в зависимости от этого ставить хирургические задачи, вырабатывать план и выбирать методы лечения. Мы наблюдали разные клинические формы и предлагаем их подразделять на следующие группы.
С.Т.Зацепин
Костная патология взрослых
Наиболее серьезным врожденным заболеванием костной системы сейчас является несовершенный остеогенез. Хотя, скорее всего, это группа патологий, обусловленных генетической предрасположенностью. Можно встретить другие названия этого заболевания: болезнь хрупких, меловых или стеклянных костей, синдром Вролика, болезнь Лобштейна, врожденная остеомаляция или периостальная дистрофия.
Патология характеризуется нарушением формирования костной ткани, вследствие чего наблюдается повышенная ломкость костей. Часто количество переломов у одного человека достигает до 100 в год. Из-за этого таких больных называют «хрустальными» или хрупкими детьми. Несовершенный остеогенез неизлечим, но в последние годы медицина облегчает страдания пациентов. И многие больные могут вести почти нормальный образ жизни.
Механизм развития
Один из 15-20 тысяч детей рождается с этой патологией. При легкой форме ребенок может нормально развиваться, а наиболее тяжелые случаи приводят к смерти уже в первый год жизни.
Несовершенный остеогенез у детей развивается из-за мутаций генов, отвечающих за синтез белков соединительной ткани. В результате снижается количество или нарушается структура коллагена 1 типа. А именно от него зависит прочность костей. Это приводит к частым переломам, деформациям скелета и другим патологиям развития.
Нарушение синтеза коллагена вызывает снижение плотности костной ткани – она имеет пористое строение и тонкий кортикальный слой. В результате этого кости, хотя растут нормально, но обладают повышенной хрупкостью.
Причины
Это заболевание относится к наследственным врожденным патологиям. Причины его – мутации генов, отвечающих за синтез белка коллагена. Болезнь чаще всего наследуется по аутосомно-доминантному типу. Это происходит, если мутации генов наблюдаются у одного из родителей. При такой форме заболевания оно протекает более благоприятно, так как переломы костей возникают после года, когда ребенок начинает ходить.
Более тяжелое течение патологии наблюдается в том случае, если наследование идет по аутосомно-рецессивному типу, то есть если оба родителя имеют мутацию генов. Встречается такая форма заболевания примерно в 5% случаев и протекает очень тяжело. Переломы могут возникать уже во время беременности, поэтому многие дети рождаются мертвыми, а 80% новорожденных не доживают до месячного возраста.

Это заболевание наследственное и передается от родителей к детям
Симптомы
Основным признаком заболевания является повышенная хрупкость костей. Переломы у таких больных случаются даже при малейшем воздействии. В самых тяжелых случаях симптомы проявляются сразу после рождения ребенка. Это случается при внутриутробной форме патологии, которая встречается примерно в 5% случаев заболевания. При этом младенцы часто получают травмы, несовместимые с жизнью, еще в период внутриутробного развития или во время родов. Они рождаются с переломами конечностей, нарушением дыхательной функции. Если ребенок с такой формой заболевания выживает, он обычно не живет больше 2 лет.
Но чаще всего встречается поздняя форма патологии. У нее более благоприятное течение. Патологическим переломам обычно подвержены трубчатые кости конечностей. Они случаются при одевании ребенка, купании, играх. Кроме хрупкости костей, наблюдаются другие деформации скелета. Чаще всего это искривление позвоночника и аномальное развитие грудной клетки. Частые патологические переломы могут приводить к неправильному сращению костей. В результате этого конечности деформируются, укорачиваются. По этим признакам на фото можно легко узнать пациента с несовершенным остеогенезом.
Подвергаются изменениям также другие органы, в функционировании которых участвует коллаген. В зависимости от типа заболевания и тяжести его течения они могут быть сильно выраженными или почти незаметными. У большинства таких больных наблюдаются характерные симптомы:
- голубоватые белки глаз;
- полупрозрачные желтоватые зубы;
- нарушение прикуса, раннее разрушение зубов;
- прогрессирующее снижение слуха;
- появление камней в почках;
- аномальная подвижность суставов, приводящая к частым вывихам;
- нарушение работы сердечных клапанов;
- атрофия мышц, слабость;
- частые носовые кровотечения;
- низкий рост.
В отличие от физического развития, интеллектуальное и психическое обычно не страдает. Дети с несовершенным остеогенезом обычно умные, эмоциональные, целеустремленные, умеют справляться с трудностями.

При тяжелых формах деформации скелета пациенты могут передвигаться только в инвалидном кресле
Виды
Эта патология имеет несколько форм, характеризующихся разными симптомами и тяжестью протекания заболевания. Принято выделять четыре типа болезни.
- Несовершенный остеогенез 1 типа – это самая легкая форма протекания болезни. Развивается она по причине недостаточной выработки коллагена. Характеризуется эта форма нетяжелыми переломами, нарушением развития зубов, тугоухостью, остеопорозом. Но во многих случаях пациенты с этой формой заболевания ведут нормальный образ жизни, так как патология не отражается на их умственном развитии. Обычно количество переломов снижается к подростковому возрасту, и только после 40 лет заболевание опять обостряется.
- При аутосомно-рецессивном наследовании часто развивается несовершенный остеогенез II типа. Это самая тяжелая форма, характеризующаяся множественными переломами костей еще во внутриутробном периоде. Новорожденные умирают от дыхательной недостаточности или кровоизлияния в мозг. При таком течении заболевания пациенты очень редко доживают до 2 лет.
- Несовершенный остеогенез III типа также развивается, когда мутация генов наблюдается у обоих родителей. Эта тяжелая форма заболевания с серьезными деформациями скелета встречается редко. Характерными признаками ее являются укороченные конечности, низкий рост, выпадение волос и очень слабые мышцы. Поэтому пациенты могут передвигаться только в инвалидном кресле.
- Несовершенный остеогенез IV типа имеет среднетяжелое течение. Он характеризуется синтезом коллагена в достаточном количестве, но этот белок имеет измененную структуру. Признаками такой формы патологии является низкий рост, деформация грудной клетки, зубов, частые переломы до 10-12 лет, снижение слуха. Но все симптомы обычно слабо выражены.

При легкой форме течения заболевания большинство пациентов могут вести нормальный образ жизни
Диагностика
Обычно диагноз ставится ребенку при рождении на основании внешних признаков и рентгенографии. Иногда делаются анализы генетического материала для определения типа заболевания. Но возможна диагностика несовершенного остеогенеза уже во время беременности. Начиная с 16 недели признаки заболевания можно обнаружить во время УЗИ. Кроме того, мутации генов выявляются при проведении генетического анализа крови.
Несмотря на то, что заболевание обнаруживается до рождения ребенка, необходимость в прерывании беременности возникает только при несовершенном остеогенезе II типа, так как эта форма заболевания протекает очень тяжело и быстро заканчивается летальным исходом.

Обнаружить патологию генов можно еще во время беременности
Несовершенный остеогенез I типа часто протекает в такой легкой форме, что пациент нормально развивается и вырастает почти здоровым. И о своем диагнозе человек может узнать только после рождения больного ребенка.
Лечение
Как и все остальные генетические заболевания, несовершенный остеогнез неизлечим. Раньше прогноз для пациентов считался очень неблагоприятным. Но современная медицина может улучшить состояние больных и позволить им вести почти нормальную жизнь. Лечение несовершенного остеогенеза направлено на замедление прогрессирования патологии и устранение ее симптомов. Сейчас полностью неблагоприятным прогнозом обладает только II тип заболевания, который 100% заканчивается смертью ребенка до 2 лет. При других формах болезни продолжительность жизни пациента и ее качество может быть не хуже, чем у здоровых людей.
Целью лечения несовершенного остеогенеза является адаптация больных к нормальной жизни, а при более тяжелом течении – обучение их навыкам самообслуживания. Поэтому для эффективного лечения важен комплексный подход.
Больной ребенок наблюдается у нескольких врачей:
- педиатр назначает медикаментозное лечение для улучшения роста и состояния костной ткани, поддерживает общее состояние здоровья;
- хирург старается предотвратить переломы с помощью ортопедической обуви, корсетов, а также следит за правильностью сращения костей;
- реабилитолог должен подобрать индивидуальную программу упражнений для адаптации ребенка к жизни, прежде всего, для нормального передвижения;
- важна также работа психолога, который помогает преодолеть страх перед переломом.
Для преодоления остеопороза – самой частой проблемы у таких пациентов – применяется медикаментозное лечение. Остановить разрежение костей можно с помощью препаратов биофосфонатов. Они тормозят синтез клеток остеокластов, которые выполняют функцию разрушения костной ткани. Начинается лечение с внутривенного введения «Памидроната». Его нужно применять каждые 2-4 месяца. Хорошие результаты наблюдаются от лечения «Ризедронатом» или золедроновой кислотой.

Очень важно для таких больных, чтобы кости после переломов срастались правильно
Иногда по показаниям назначают гормон роста. Он помогает ускорить рост трубчатых костей и улучшить обменные процессы в костной ткани. Также применяются соли магния, фосфора и калия, препараты кальция и витамин Д, «Соматотропин», гормоны паращитовидных желез. Показано проведение электрофореза с солями кальция, УФО, магнитотерапии, индуктотермии, массажа.
Очень важна для таких пациентов адекватная физическая нагрузка. Многие дети после болезненных переломов боятся движения и предпочитают сидеть или лежать. Это приводит к атрофии мышц. Кроме того, у обездвиженных больных развивается гипокинетический остеопороз, который еще больше разрушает костную ткань. Поэтому одной из основных задач лечения несовершенного остеогенеза является обучение пациентов безопасным способам передвижения и специальным упражнениям.
Если деформации скелета сильно мешают пациенту двигаться, требуется хирургическое лечение. Операция довольно сложная, так как кость разрезают и сопоставляют ее так, чтобы она приняла правильную форму. После этого ее укрепляют штифтом или специальным гибким теном, который вводится в костный канал.

Для больного ребенка очень важно создать правильную развивающую среду, побуждающую его двигаться
Для благоприятного течения заболевания важная роль отводится родителям. Потому что самое главное для «хрустального» ребенка – это научиться жить со своей проблемой, вести себя так, чтобы предотвратить переломы. Задача родителей – правильно ухаживать за больным ребенком, следить за регулярным прохождением реабилитационного лечения. Важно побуждать малыша самостоятельно двигаться, учить его обслуживать себя. Для этого нужно использовать в доме вспомогательные приспособления, например, поручни, ступеньки, специальные сидения. Очень полезно больному ребенку заниматься плаванием, танцами, музыкой, ручным творчеством.
При правильном подходе к лечению дети с несовершенным остеогенезом нормально развиваются. Они часто даже более способные и талантливые, так как отличаются целеустремленностью и умением преодолевать трудности.
Осложнения
Основным симптомом заболевания является повышенная ломкость костей. Но из-за того, что нарушено костеобразование, могут развиваться различные осложнения. Чаще всего это деформации конечностей, искривление позвоночника и грудной клетки. Грудь может приобрести бочкообразную форму, развивается сильная сутулость. Конечности часто укорочены, искривлены.
Кроме того, распространенным осложнением заболевания является нарушение прикуса, быстрое разрушение зубов. Из-за разрежения костной ткани страдает также слух. У большинства пациентов с возрастом развивается тугоухость или глухота. А деформации грудной клетки приводят к болезням дыхательной системы.
Современная медицина добилась того, что с таким серьезным и неизлечимым заболеванием человек может прожить нормальную жизнь. Начиная с 2012 года «хрустальные» дети получают лечение на благотворительной основе. Существуют международные фонды и группы, в которых объединены родители. Там они могут получить совет, помощь и поддержку.
3021 0
Заболевания костей и суставов могут поражать даже специалистов медицинской деятельности, которые имеют огромный опыт в своей сфере и достигли определенного уровня мастерства.
Но даже опытные врачи приходят в ужас, когда видят новорожденного «хрустального» человека.
Понятие и статистика
Несовершенный остеогенез или «болезнь хрустального человека» представляет собой серьезное нарушение внутриутробного развития костей и суставов.
Заболевание характеризуется повышенной хрупкостью костей и суставов скелета у человека. Объяснение представленному заболеванию можно найти в нехватке коллагена в организме больного, либо несоответствия показателям нормы описываемого важного белка в строении костного скелета.
Заболевание относится к генетическим проявлениям и возникает у малышей, чьи родители также страдают патологией.
В редких случаях болезнь диагностируется у детей совершенно здоровых родителей и родственников. Такие особенности объясняются спонтанной мутацией.
Статистика заболевания может привести беременных женщин в паническое состояние, поскольку показатель фиксированных случаев составляет 1 новорожденный ребенок на 15 тысяч всех беременностей.
Не следует поддаваться эмоциям, поскольку современные медицинские исследования и методы лечения могут привести к положительным результатам оздоровления больного ребенка.
Причины заболевания
Как уже было оговорено выше описываемое заболевание является следствием наследственной мутации генов коллагена, что приводит к нарушению его строения или нехватке в организме.
Также на проявление «болезни хрустального человека» влияет нехватка синтезированного коллагена.
Такой вид заболевания протекает в более легкой форме и характеризуется повышенной степенью ломкости костей, что приводит к частым переломам у больного человека. После пубертатного периода количество переломов значительно уменьшается, а в зрелом возрасте все повторяется.
Объяснить причины спонтанной мутации специалисты не могут. Единственное, что можно посоветовать беременным женщинам – это внимательно относиться к своему здоровью и проходить регулярное обследование в период беременности.
Не употребляйте алкоголь и откажитесь от курения во время внутриутробного развития человека.
Типы и симптомы заболевания
Несовершенный остеогенез имеет несколько форм проявления и развития, что характеризуется отличительными симптомами и структурой костей больного человека.
Тип I – слабая форма
Количество страдающих именно этим типом составляют примерно 50% всех выявленных случаев. Как уже упоминалось, больные подвержены частым переломам костей и вывихам суставов.
Риск переломов снижается после 10 лет жизни, но уже после 40 летнего возраста больной возвращается в группу риска.
При первом типе происходят определенные изменения в аорте, в результате чего можно наблюдать частые носовые кровотечения.
Тип II – перинатально-летальный
Эта форма проявления несовершенного остеогенеза характеризуется частой смертью плода еще в период беременности женщины. В ином случае происходят преждевременные роды на малых сроках беременности. Здесь также выделяют три группы:
- Группа А – фиксируются повреждения головы еще на этапе внутриутробного развития. Дети рождаются ростом всего 20-30 см. Ярко выражены нарушения мозговой активности и дыхательной системы. Новорожденные либо рождаются мертвыми, либо умирают в первые несколько дней (в редких случаях умирают в конце первого месяца жизни). Смерть ребенка вызвана многочисленными переломами.
- Группа Б – признаки проявления заболевания те же, что и у группы А, за исключением нормального развития дыхательной си или с небольшими отклонениями от норм. Такие новорожденные могут прожить несколько лет. У них отмечается укороченность всех трубчатых костей.
- Группа В – диагностируется очень редко. Новорожденные умирают в первые дни жизни или уже рождаются мертвыми. Отмечается истончение трубчатых костей и отсутствие оссификации черепа.
Тип III — нарушение роста костей
Тип III встречается крайне редко и характеризуется нарушением роста костей.
Тело новорожденного при малом росте может иметь нормальный вес. Также диагностируется нарушение кровообращения, что в большинстве случаев становится причиной смерти. Фиксируют переломы костей во время родов.
Тип IV- нарушения в росте скелета
Тип IV характеризуется наличием нарушений скелета. Через несколько лет у больного возникают мозоли костей, а количество переломов уменьшается. После 30 отмечается нарушение слуха.
Выявить тип или группу патологии помогает комплексная диагностика, которая проводится сразу же после рождения малыша.
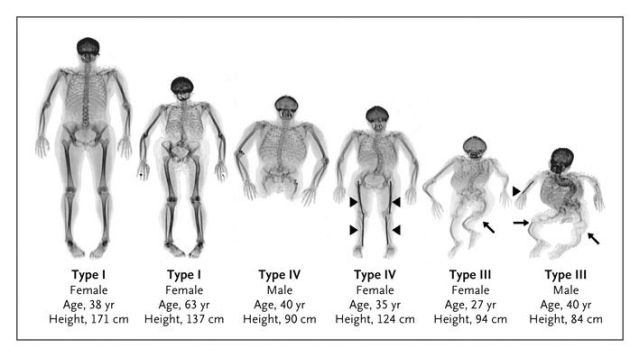
На фото несовершенный остеогенез четырех типов
Диагностика заболевания
Диагностика происходит в два этапа. Следует также отметить, что существует внутриутробная диагностика. Здесь используется ультразвуковое исследование.
При подозрениях на наличие нарушений структуры коллагена проводят дополнительный ряд химических обследований с забором амниотической жидкости и эпителиальных тканей у беременной женщины.
Сразу же после рождения больного малыша проводят ряд инструментальных исследований, где выделяют:
- рентген – с помощью снимка можно выявить имеющиеся переломы;
- денситометрия – осуществляется исследование минеральной полости костной ткани;
- производят забор костной ткани для биопсии.
Помимо инструментальных проводятся и лабораторные исследования:
- на основе крови выявляются нарушения в структуре ДНК;
- проводят сдачу анализов на диагностику коллагена;
- производят несколько анализов на основе биопсии кожи.
На основании проведенной диагностики специалисты составляют схему подходящего лечения.
Лечение патологии
 Лечение несовершенного остеогенеза происходит несколькими методами, в числе которых выделяют медикаментозный способ, основанный на применении больным препаратов для увеличения плотности костей с целью уменьшить количество переломов.
Лечение несовершенного остеогенеза происходит несколькими методами, в числе которых выделяют медикаментозный способ, основанный на применении больным препаратов для увеличения плотности костей с целью уменьшить количество переломов.
В состав лекарственных препаратов должны входить кальций, витамин D, соли калия, магния и прочие полезные химические вещества.
Помимо медикаментозной терапии используют физиотерапию и лечебную гимнастику. Также в основе лечения состоит психотерапия, которую проходят родители больного малыша и прочие родственники.
Задача психотерапии объяснить основные правила и методы для дальнейшего обучения малыша поведению в обществе, чтобы он самостоятельно мог избегать ситуаций, которые могут повлечь перелом.
Осложнения и прогноз
Предупредить и обезопасить своего ребенка от развития патологии невозможно, но при своевременном лечении и выполнении всех советов и рекомендаций, высказанных врачом, новорожденные малыши могут вырасти и реализоваться в жизни, поскольку характерных психологических или умственных отклонений у детей не выявляется.
Осложнения же могут спровоцировать полученные при падении, ведь для «хрустального» человека небольшой удар мячом по спине во время игры в футбол может закончиться переломом позвоночника.
Именно поэтому жизнь хрустальных людей нельзя сравнивать с жизнедеятельностью здоровых.
Они должны оберегать себя от малейших травм и ушибов, любое падение может привести к перелому ноги и последующему нахождению в инвалидном кресле.
Если родители примут своего ребенка и начнут соответствующие действия для его выздоровления, можно добиться хороших результатов и положительной динамики.
Полного выздоровления, как правило, не последует, но благодаря современному техническому прогрессу можно существенно облегчить жизнь больного.
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Клинические протоколы МЗ РК - 2016
Незавершенный остеогенез (Q78.0)
Орфанные заболевания
Общая информация
Краткое описание
---Одобрено
Объединенной комиссией по качеству медицинских услуг
Министерства здравоохранения и социального развития Республики Казахстан
от «29» сентября 2016 года
Протокол №11
Несовершенный остеогенез (НО, Болезнь хрупких костей, болезнь стеклянных костей, болезнь Лобштейна-Вролика, остеопсатироз, болезнь Порак и Дуранте) - гетерогенная группа генетических расстройств характеризующихся повышенной хрупкостью костей, снижением массы костной ткани, и склонностью к переломам костей различной степени тяжести .
Соотношение кодов МКБ-10 и МКБ-9:
| МКБ-10 | МКБ-9 |
| Q.78.0 Незавершенный остеогенез |
33.34 Торакопластика; 77.22 Клиновидная остеотомия плечевой кости; 77.27 Клиновидная остеотомия большеберцовой и малоберцовой кости; 78.19 Применение внешнего фиксирующего устройства на прочие кости при заболеваниях, требующих этапной коррекции; 79.19 Закрытая репозиция костных обломков другой уточненной кости с внутренней фиксацией 79.31 Открытая репозиция костных обломков плечевой кости с внутренней фиксацией; 79.311 Открытая репозиция костных отломков плечевой кости с внутренней фиксацией блокирующим интрамедуллярным остеосинтезом; 79.32 Открытая репозиция костных обломков лучевой и локтевой кости с внутренней фиксацией; 79.321 Открытая репозиция костных отломков лучевой и локтевой кости с внутренней фиксацией блокирующим интрамедуллярным остеосинтезом; 79.35 Открытая репозиция костных обломков бедренной кости с внутренней фиксацией; 79.351 Открытая репозиция костных отломков бедренной кости с внутренней фиксацией блокирующим интрамедуллярным остеосинтезом; 79.36 Открытая репозиция костных обломков большеберцовой и малоберцовой костей с внутренней фиксацией; 79.39 Открытая репозиция костных обломков другой уточненной кости с внутренней фиксацией; 79.391 Открытая репозиция костных отломков другой уточненной кости с внутренней фиксацией блокирующим экстрамедуллярным имплантом; 84.991 Наложение аппарата для компрессионно-дистракционного остеосинтеза; |
Дата разработки/пересмотра протокола: 2016 год.
Пользователи протокола : врачи общей практики, педиатры, детские хирурги, травматолог-ортопеды, эндокринологи, генетики, медицинские реабилитологи, реаниматологи.
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С |
Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+). Результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
Классификация
Классификация
Основным клиническим проявлением всех типов НО является хрупкость костей, которая проявляется в виде спонтанных переломов. В настоящее время широко применяют классификацию, которая базируется на данных клинического и рентгенологического обследования пациента и позволяет выделить четыре генетических типа заболевания (Таблица 1).
Таблица 1.

На сегодня выделено еще четыре типа НО (V, VI, VII, VIII), которые не связаны с патологией коллагена I типа и пока не внесены в Международную классификацию остеохондропатий. В связи с этим, представлена другая классификация несовершенного остеогенеза, которая имеет восемь клинических различных типов НО (Таблица 2) .
Таблица 2. Классификация НО

Диагностика (амбулатория)
ДИАГНОСТИКА НА АМБУЛАТОРНОМ УРОВНЕ
Диагностические критерии (УД - В)
Жалобы:
· повышенная ломкость трубчатых костей;
· деформация и укорочение конечностей;
· искривление позвоночника;
· серо-синие склеры глаз;
· деформация грудной клетки и ребер;
· хрупкость и ломкость зубов;
· снижение слуха;
· отставание физического развития;
· слабость мышц.
Анамнез:
· наличие заболевания у одного из родителей или у дальних родственников;
· наличие многочисленных переломов;
· клинически установленный диагноз «Несовершенный остеогенез».
Физикальное обследование:
· Повышенная ломкость костей.
· Изменение формы и укорочение костей в результате неправильного сращения переломов.
· Деформация (изменение формы) грудной клетки.
· Мягкие кости черепа.
· Серо-синяя склера (белок) глаза из-за недоразвития его соединительной ткани и просвечивания внутренней оболочки, содержащей пигмент (красящее вещество).
· Позднее прорезывание зубов у детей (позже 1,5 лет), крошение зубов; цвет зубов желтый — “ янтарные зубы”.
· Слаборазвитые мышцы (дряблые, значительно уменьшены в объеме).
· Часто возникают паховые, пупочные грыжи.
· Слабость связочного аппарата сустава.
· Снижение слуха из-за прогрессирующего разрастания соединительной ткани между мелкими косточками (молоточек, наковальня, стремечко) полости среднего уха.
· Отставание в физическом развитии.
· Низкий рост.
Лабораторные исследования: нет.
(УД - В):
Диагностический алгоритм :
Рисунок 1. Диагностический алгоритм

Диагностика (стационар)
ДИАГНОСТИКА НА СТАЦИОНАРНОМ УРОВНЕ
Диагностические критерии:
Диагностический алгоритм: смотрите амбулаторный уровень.
Лабораторные исследования: нет.
Инструментальные исследования
(УД - В):
· Рентгенологическое исследование - основной клинический признак распространенный остеопороз (уменьшение плотности кости, способствующее снижению прочности) всего скелета.
· Компьютерная томография - отмечается множественная многоплоскостная деформация трубчатых костей, системный остеопороз. Корковый слой истончен, местами, где надкостница в пределах диафиза непосредственно прилегает к губчатому веществу, компактная краевая каемка отсутствует. Костномозговой канал в связи с этим эксцентрически увеличен в диаметре и местами неровен. Губчатая структура разрежена и имеет широко- петлистый, сетчатый, а иногда неправильный хаотический рисунок, отдельные трабекулы едва выступают.
Перечень основных диагностических мероприятий:
· рентгенологическое исследование;
· компьютерная томография.
Перечень дополнительных диагностических мероприятий:
· Денситометрическое исследование - отмечается снижение уровня минеральной плотности костной ткани. Низкая костная минеральная плотность по отношению к хронологическому возрасту может быть Z-критерии ≤ -2.0 SD.
Дифференциальный диагноз
1)
Дифференциальный диагноз и обоснование дополнительных исследований
Таблица 3. Дифференциальная диагностика НО
| Признак | НО | ЮИО | ГФФ | Синдром псевдоглиомы |
| Переломы и деформация костей | + | + | + | + |
| Серо-синие склеры глаз | + | - | - | - |
| Нарушение прорезывания зубов | + |
- |
+ | - |
| Семейный анамнез |
+ |
- | - | + |
| Нарушение слуха | + | - | - | - |
| Нарушение когнитивной функции | - | - | - | + |
| Рентгенологические изменения | деформация трубчатых костей | деформация на уровне метафизов | рахитоподобные нарушения | признаки остеопороза |
| Денситометрия, снижение МПКТ | + | + | - | + |
| Патология соединительной ткани | + | - | - | + |
| Молекулярные дефекты | + | - | + | + |
| Слепота | - | - | - | + |
| ЩФ в крови | n | n | ↓ | n |
| Фосфоэтаноламин в моче | n | n | n |
Лечение за рубежом
Пройти лечение в Корее, Израиле, Германии, США
Получить консультацию по медтуризму
Лечение
Лечение (амбулатория)
ЛЕЧЕНИЕ НА АМБУЛАТОРНОМ УРОВНЕ
Тактика лечения
:
Лечение, предписанное в настоящее время, включает:
· адаптацию поведения и образа жизни во избежание ситуаций, которые могут повлечь за собой перелом;
· ортезирование;
· коррекцию осанки;
· консервативное лечение, включающую водные процедуры и физическую активность;
· специальное оборудование, в том числе обеспечивающее передвижение;
· контроль веса;
· оральное и внутривенное применение бисфосфонатов.
Немедикаментозное лечение:
Адаптация поведения и образа жизни. Проведение щадящих ЛФК и физиопроцедур.
Медикаментозное лечение
Лекарственные препараты типа бифосфонатов (1,2,3 поколений) представляют собой мощные ингибиторы резорбции кости (вещество, предотвращающее разрущение костной ткани).
· Памидроновая кислота, концентрат для приготовления раствора для инфузий 3 мг/мл, во флаконах - 30мг/10мл или 90мг/30мл. Фармакологическое действие - ингибирующее костную резорбцию.
Дозы:
До 2 лет - 0.5 мг/кг/день, 1 раз в 2 мес.
2.1 -3 г. - 0.75 мг/кг/день, 1 раз в 3 мес.
>3 л. - 1 мг/кг/день, 1 раз в 4 мес.
Не более 60 мг/день, в среднем 9 мг/кг в год.
Таблица 4. Схема применения памидроновой кислоты у детей (Plotkinet H. et al., 2000)

Рисунок 2. Алгоритм назначения памидроновой кислоты у пациентов с несовершенным остеогенезом

Перечень основных лекарственных препаратов:
:
· консультация генетика - для верификации типа НО и прогнозирования вероятности заболевания при повторных беременностях;
· консультация оториноларинголога - при наличии тугоухости;
· консультация стоматолога - при несовершенном дентиногенезе, дисплазии зубов, кариесе и др.;
· консультация педиатра - при наличии пневмонии, анемии, снижении индекса массы тела и других состояний.
· консультация эндокринолога - при наличии низкорослости, нанизма и других состояний.
Профилактические мероприятия
:
· медико-генетическая консультация;
· беседа с родителями о высоком риске рождения больного ребенка, а также о возможности мертворождения при втором типе НО, а также летального исхода от множественных переломов и др.;
· перинатальная диагностика;
· медико-социальная реабилитация;
· устранение факторов риска (механической травматизации, внешнего и других видов воздействия);
· лечение сопутствующей патологии;
· использование пневмошин и ортопедических изделий;
· санаторно-курортное лечение.
Мониторинг состояния пациента:
· диспансерный учет по месту жительства у детского травматолога-ортопеда, хирурга, педиатра;
· наблюдение и лечение у смежных специалистов;
Индикаторы эффективности лечения:
· уменьшение количество переломов и слизистых оболочек;
· коррекция деформаций конечностей;
· улучшение моторных функций;
· улучшение общего состояния.
Лечение (скорая помощь)
ДИАГНОСТИКА И ЛЕЧЕНИЕ НА ЭТАПЕ СКОРОЙ НЕОТЛОЖНОЙ ПОМОЩИ
Диагностические мероприятия: нет.
Медикаментозное лечение:
Медикаментозное лечение, оказываемое на этапе скорой неотложной помощи (смотреть КП по соответствующим нозологиям по переломам):
· иммобилизация конечности;
· купирование болевого синдрома.
Лечение (стационар)
ЛЕЧЕНИЕ НА СТАЦИОНАРНОМ УРОВНЕ
Тактика лечения:
лечение на стационарном уровне по экстренной госпитализации согласно клиническим протоколам по соответствующим нозологиям по переломам.
При плановой госпитализации с целью предоперационной подготовки может включать лечение по амбулаторному уровню.
Немедикаментозное лечение: смотрите амбулаторный уровень.
Медикаментозное лечение: смотрите амбулаторный уровень.
Симптоматическая терапия включает наркотические и ненаркотические анальгетики в послеоперационном периоде (трамадол, парацетамол, ибупрофен и др.), антибактериальные средства для профилактики и лечения инфекционных осложнений (антибиотики- пенициллины, цефалоспорины, аминогликозиды, карбапенемы др.), противогрибковые средства для профилактики и лечения микозов (флуконазол, каспофунгин и др.).
Хирургическое вмешательство
:
· Клиновидная остеотомия плечевой кости;
· Клиновидная остеотомия большеберцовой и малоберцовой кости;
· Применение внешнего фиксирующего устройства на прочие кости при заболеваниях, требующих этапной коррекции;
· Закрытая репозиция костных обломков другой уточненной кости с внутренней фиксацией
· Открытая репозиция костных обломков плечевой кости с внутренней фиксацией;
· Открытая репозиция костных отломков плечевой кости с внутренней фиксацией блокирующим интрамедуллярным остеосинтезом;
· Открытая репозиция костных обломков лучевой и локтевой кости с внутренней фиксацией;
· Открытая репозиция костных отломков лучевой и локтевой кости с внутренней фиксацией блокирующим интрамедуллярным остеосинтезом;
· Открытая репозиция костных обломков бедренной кости с внутренней фиксацией;
· Открытая репозиция костных отломков бедренной кости с внутренней фиксацией блокирующим интрамедуллярным остеосинтезом;
· Открытая репозиция костных обломков большеберцовой и малоберцовой костей с внутренней фиксацией;
· Открытая репозиция костных обломков другой уточненной кости с внутренней фиксацией;
· Открытая репозиция костных отломков другой уточненной кости с внутренней фиксацией блокирующим экстрамедуллярным имплантом;
· Наложение аппарата для компрессионно-дистракционного остеосинтеза;
· Торакопластика.
Другие виды лечения
:
· психологический тренинг;
· обучение пациента.
Показания для консультации специалистов
:
· консультация генетика - для верификации типа НО и прогнозирования вероятности заболевания при повторных беременностях;
· консультация оториноларинголога- при наличии тугоухости;
· консультация стоматолога - при несовершенном дентиногенезе, дисплазии зубов, кариесе и др.;
· консультация педиатра - при наличии пневмонии, анемии, снижении индекса массы тела и других состояний.
· консультация эндокринолога - при наличии низкорослости, нанизма и других состояний.
Показания для перевода в отделение интенсивной терапии и реанимации:
· травматический шок II-III степени;
· первые сутки после объемной операции.
Индикаторы эффективности лечения
:
· уменьшение количество переломов и слизистых оболочек;
· коррекция деформаций конечностей;
· улучшение моторных функций;
· улучшение общего состояния.
Дальнейшее ведение
:
· диспансерный учет по месту жительства у детского травматолога-ортопеда, хирурга, педиатра;
· наблюдение и лечение у смежных специалистов.
Госпитализация
Показания для плановой госпитализации:
· наличие деформации конечностей.
Показания для экстренной госпитализации:
· при переломах крупных трубчатых костей, со смещением требующих их остеосинтез.
Информация
Источники и литература
- Протоколы заседаний Объединенной комиссии по качеству медицинских услуг МЗСР РК, 2016
- 1) Поворознюк В.В., Гречанина Е.Я, Балацкая Н.И., Вайда В.М.. Несовершенный остеогенез: патогенез, классификация, клиническая картина, лечение. Ортопедия, травматология и протезирование – 2009, №4:110-117c. 2) Попков А.В.. Осипенко А.В. Регенерация тканей при удлинении конечностей: Руководство для врачей. – М.: ГЭОТАР-Медиа, 2008. – 240с. 3) Сатжанов А.Б. Усовершенствование хирургического лечения деформаций нижних конечностей у детей с несовершенным остеогенезом // Диссертация – 2016г., 67с. 4) Astrom E., Soderhall S. Beneficial effect of bisphosphonate during five years of treatment of severe osteogenesis imperfecta. Acta Paediatr (NORWAY). 1998, 87 (1): 64-8 5) Anum E.A., Hill L.D., Pandya A., Strauss J.F. Connective Tissue and Related Disorders and Preterm Birth: Clues to Genes Contributing to Prematurity // Placenta. 2009. - Vol. 30. - P. 207-215. 6) Astrom A. Beneficial effect of long term intravenous bisphosphonate treatment of osteogenesis imperfecta [Текст] / A. Astrom, S. Soderhall // Arch. Dis. Child. - 2002. - Vol. 86. - P. 356¬364. 7) Baldridge D., Schwarze U., Morello R., Lennington J., Bertin T. K., Pace J.M., et.al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta // Hum Mutat. 2008. - P. 1435-1442. 8) Barnes A.M., Carter E.M., Cabral W.A. et al. Lack of Cyclophilin B in Osteogenesis Imperfecta with Normal Collagen Folding // The new england journal of medicine. 2010. -V.362. - P.521-528. 9) Barnes A.M., Chang W., Morello R., Cabral W.A., Weis M., Eyre D.R., Leikin S. et al. Deficiency of Cartilage-Associated Protein in Recessive Lethal Osteogenesis Imperfecta // N Engl J Med. 2006. - Vol. 355. - P. 2757-64. 10) Ben Amor I.Mouna, Glorieux Francis H., Rauch Frank. Genotype-Phenotype Correlations in Autosomal Dominant Osteogenesis Imperfecta // Journal of Osteoporosis. 2011. - P. 9. 11) Bembi B., Parma A., Bottega M., Ceschel S., Zanatta M., Martini C., Ciana G. Intravenous pamidronate treatment in osteogenesis imperfecta. J Pediatr (UNITED STATES). 1997, 131 (4):622-5 12) Byers P.H. Oseogenesis imperfecta: perspective and opportunities // Curr Opin Pediatr. 2000. - P. 603-609. 13) Breslau-Siderius E.J., Engelbert R.H., Pals G., Van der Sluijs J*.A. Bruck syndrome: a rare combination of bone fragility and multiple congenitaljoint contractures // J Pediatr Orthop B. 1998. - Vol. 7. - P. 35-38. » 14) Glorieux F.H. et al. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta/ // N. Engl. J. Med. - 1998. - Vol. 339. - P. 947¬952. 15) Guilot M., Ekcart P.,Desrosieres H.,AmiourM., al-Jazauri Z. . Arch Pediatr. 2001, 8:172-75 16) Byers P.H., Steiner R.D. Osteogenesis imperfecta // Annu Rev Med. -1992.-Vol. 43.-P. 269-282. 17) Chan TF, Poon A, Basu A, Addleman NR, Chen J, Phong A, Byers PH, Klein TE, Kwok PY. Natural variation in four human collagen genes across an ethnically diverse population // Genomics. - 2008. Vol. 91(4). -P. 307-14. 18) Cheung M.S., Francis H. Gloireiux, Frank Rauch. Intravenous pamidronate in osteogenesis imperfecta type VII [Текст] / Moira S. Cheung, Francis H. Gloireiux, Frank Rauch // Calcified tissue international. - 2009. - Vol. 84. - P. 203¬209. 19) Cheung M.S., Glorieux F.H. Osteogenesis Imperfecta: Update on presentation and management // Rev Endocr Metab Disord. - 2008. Vol. 9.-P. 153-160 20) Cinman N. Osteogenesis imperfecta. A life not so fragile // Lancet 358 Suppl: S46.-2001. 21) Cooper C., Dennison E.M., Leufkens H.G., Bishop N.,. Van Staa T.P: Epidemiology of childhood fractures in Britain: a study using the general" practice research database // J Bone Miner Res.- 2004. -Vol. 19.- P.-1976-1981. 22) Crabtree N.J., W. Hogler, N.J. Shaw Longitudinal changes in untreated children with osteogenesis imperfecta / // Bone. - 2009. - Vol. 45. - P. 75. 23) Cubert R:, Cheng E.Y., Mack S., Pepin M:G., Byers P.H. Osteogenesis imperfecta: mode of delivery and neonatal outcome // Obset Gynecol. 2001. Vol. 97(1).-P. 66-9. 24) Dimitri P. Changes in body composition following 3 years of pamidronate therapy in osteogenesis imperfecta / P. Dimitri, J. Crook, N. Bishop // Bone. - 2007. - Vol. 40. - P. 22¬89. 25) Devogelaer J.P., Nagant de Deuxchaisnes C. Use of pamidronate in chronic and acute bone loss conditions. Medicina (B Aires) (ARGENTINA). 1997, 57, Suppl 1:101-8 26) Elazabi A. Spinal bone mineral density in children and adolescents treated with cyclical intravenous pamidronate [Текст] / A. Elazabi, J.E. Adams, M.Z. Mughal // Bone. - 2009. - Vol. 45. - P. 104. 27) Epstein M.P., Satten G.A. Inference on haplotype effects in case-control studies using unphased genotype data // Am. J. Hum. Genet. 2003. - Vol.73.-P. 1316-1329. 28) Fleisch H. Bisphosponates: mechanisms of action // Endocr Rev. - 1998. -Vol. 19.-P. 80-100. 29) Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. -2011. -Vol. 7(9). P. 54057. 30) Fraser R.D.B., MacRae T.P. and Suzuki E., Chain conformation in the collagen molecule // J. Mol. Biol. 1979. - Vol. 129. - P. 463-481. 31) Fujiwara I., Ogawa E., Igarashi Y., Ohba M., Asanuma A. Intravenous pamidronate treatment in osteogenesis imperfecta . Eur J Pediatr (GERMANY). 1998, 157 (3): 261-2 32) Gajko-Galicka A. Mutations in type I collagen genes resulting in osteogenesis imperfecta in humans // Acta Biochimica Polonica.- 2002. -Vol. 49.-No. 2.-P. 433-441. 33) Kuurila, K., Kaitila, I., Johansson, R. & Grenman, R. Hearing loss in Finnish adults with osteogenesis imperfecta: a nationwide survey I I Ann. Otol. Rhinol. Laryngol. 2002. - Vol. 111.- P. 939-946. 34) Engelbert R.H:, Pruijs H.E.-, Beemer F.A., Helders P.J. Osteogenesis imperfecta in childhood: treatment strategies // Arch Phys Med Rehabil.1998. Vol. 79. - P. 1590-1594. 35) Glorieux F.H., Bishop N.J., Plotkin H., Chabot G., Lanoue G., Travers R. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta // N Engl J Med. 1998. - Vol. 339. - P. 947-952. 36) Glorieux F.H., Rauch F., Plotkin H., Ward L., Travers R., Roughley P., et al. Type V osteogenesis imperfecta; a new form of brittle bone disease // J Bone Miner Res. 2000. - Vol. 15. - P. 1650-1658 37) Glorieux F.H., Ward L.M., Rauch F., Lalic L., Roughley P.J., Travers R. Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect // J Bone Miner Res. 2002. - Vol. 17. - P. 30-38. 38) Gonzales E., PaviaC., Ros J.,Villaronga M.,Valls C., Escola J. Efficacy of low dose schedule pamidronate infusion in children with osteogenesis imperfecta. J Pediatr Endocrinol Metab. 2001, 14:529-33 39) Falk M.J., Heeger S., Lynch K.A. et al. Intravenous bisphosphonate therapy in children with osteogenesis imperfecta. Pediatrics. [Текст] / // Clinical Trial. Journal Article. - 2003. - Vol. 111 (3). - P. 573¬578. 40) Intravenous pamidronate treatment on children with moderate to severe Osteogenesis imperfecta started less than 36 month of age [Текст] / Alcausin M.B., Ault J., Pacey V. et al. // Bone. - 2009. - Vol. 45. - P. 90. 41) Land C. Effects of cyclical intravenous pamidronate treatment [Текст] / C. Land, F. Rauch, R. Travers, F.H. Glorieux // Bone. - 2007. - Vol. 40 - P. 638¬644. 42) Rauch F. The effects of intravenous pamidronate on bone tissue of children and adolescents with osteogenesis imperfecta / F. Rauch, R. Travers, H. Plotkin, F.H. Glorieux // J. Clin. Invest. - 2002. - Vol. 110. - P. 1293¬1299. 43) Landin LA. Fracture patterns in children. Analysis of 8,682 fractures with special reference to incidence, etiology, and secular changes in a Swedish urban population 1950-1979 // Acta Orthop Scand Suppl. 1983.-Vol. 202.-P. 1-109. 44) Lee Y.S., Low S.L. Lim L.A. Loke K.Y. Cyclic pamidronate infusion improves bone mineralization and reduce fracture incidence in osteogenesis imperfecta. Eur J Pediatr. 2001,160:641-4 45) Long¬bone changes after pamidronate discontinuation in children and adolescents with osteogenesis imperfecta / Frank Rauch, Sylvie Cornibert, Moira Cheung, Francis H. Glorieux // Bone. - 2007. - Vol. 40. - P. 821¬827. 46) Lindsay R. Modiling the benefits of pamidronate in children with osteogenesis imperfecta. J Clin Invest. 2002,110:1239-41 47) Marini J.C. Evaluation of growth hormone axis and responsiveness to growth stimulation of short children with osteogenesis imperfecta / J.C. Marini, S. Bordenick, G. Heavner // Am. J. Med. Genet. - 1993. - Vol. 45. - P. 261¬264. 48) Materials 6th International conference on osteogenesis imperfecta. 19-21 September 1996, Zeist, Netherlands 49) McCarthy E.A.,Raggio C.L., Hossak M.D., Miller E.A.,Boskey A.L.,Camacho N.P. Alendronate treatment for infants with osteogenesis imperfecta: demonstration of efficacy in a mouse model. Pediatr Res. 2002,52:660-70 50) Paley D. Principles of deformity correction. – Berlin–Heidelberg: Springer-Verlag, 2002. – 806 p. 51) Paley D., Maar D.C. Ilizarov bone transport treatment for tibial defects // J. Orthop. Trauma. – 2000. – Vol. 14, № 2. – P. 76–85. 52) Pamidronate treatment of severe osteogenesis imperfecta in children under 3 years of age / Horacio Plotkin, Frank Rauch, Nicholas J. Bishop // J. Clin. Endocrinol. Metab. - 2000. - Vol. 85. - P. 1846¬1850. 53) Plotkin H., Rauch F., Bishop N.J.,Montpetit K., Ruck-Gibis J., Travers R., Glorieux F.H. Pamidronate treatment of severe osteogenesis imperfecta in children under 3 years of age. J Pediatr Endocrinol Metab. 2000, 85:1846-50 54) Rauch F. Osteogenesis imperfecta [Текст] / F. Rauch, F.H. Glorieux // Lancet. - 2004. - Vol. 363. - P. 1377¬1385. 55) Rauch F. The effects of intravenous pamidronate on the bone tissue of children and adolescents with osteogenesis imperfecta / Frank Rauch, Rose Travers, Horacio Plotkin, Francis H. Glorieux // J. Clin. Invest. - 2002. - Vol. 110. - P. 1293¬1299. 56) Rauch F., Gloneux E.H: Osteogenesis imperfecta // Lancet. 2004". - Vol. 363.-P. 1377-85. 57) Rauch F., Plotkin H:, Zeitlin L., Glorieux F.H. Bone mass, size and density in children and: adolescents with;, osteogenesis imperfecta: Effect of intravenous Pamidronate therapy // J Bone Miner Res. 2003. - Vol. 18.-P. 610-614. 58) RauchF:, Traverse R:, Norman M.E., Taylor A., Parfitt A.M:, Glorieux F.Hi Deficient bone formation, in idiopathic juvenile osteoporosis: a histomorphometric study of cancellous iliac bone // J Bone Miner Res. - 2000. Vol. 15. - P. 957-63. 59) Respiratory distress with pamidronate treatment in infants with severe osteogenesis imperfecta [Текст] / Craig F. Munns, Frank Rauch, Richard J. Mier, Francis H. Glorieux // Bone. - 2004. - Vol. 35. - P. 231¬234. 60) Ripamonti U. Smart biomaterials with intrinsic osteoinductivity: geometric control of bone differentiation // Bone Engineering; Ed. J.M. Davies. – Toronto: Em Squared Inc., 2000. 61) Rivera E.M., Araiza M., Brostow W. et al. Synthesis of hydroxyapatite from eggshells // Matter. Lett. – 1999. – Vol. 41. – P. 128–134. 62) Schwartz S. Bisphosphonates, Osteonecrosis, Osteogenesis Imperfecta and Dental Extractions: A Case Series [Текст] / Clara Joseph, Deborah Iera, Duy¬Dat Vu // JCDA. - 2008. - Vol. 74 (6). - P. 538¬542. 63) Suffering from osteogenesis imperfecta [Текст] / Devogelaer J.P., Malghem J., Maldague B., Nagant de Deuxchaisnes // Skel. Radiol. C. - 1987. - Vol. 16. - P. 360¬363. 64) Transplantation of unrelated placental blood cells in children with high¬risk sickle cell disease / Adamkiewicz T.V., Mehta P.S., Boyer M.W. et al. [Текст] // Bone Marrow Transplant. - 2004. - 34 (5). - Р. 405. 65) Tripon P., Dalzotto G., Poichotte A. et al. Reconstruction of post-traumatic diaphyseal bone loss by segmental bone transfer // Ann. Chir. Plast. Esthet. – 2000. – Vol. 45, № 3. – P. 336–345. 66) Vertebral deformities in children with osteogenesis imperfecta: effects of intravenous pamidronate and neridronate treatment [Текст] / R. Beccarda, O. Semlera, C. Landb et al. // Bone. - 2009. - Vol. 45. - P. 59¬111. 67) Land Cristof, Frank Rauch, Craig F. Munns et al. Vertebral morphometry in children and adolescents with osteogenesis imperfecta: Effect of intravenous pamidronate treatment // Bone. - 2006. - Vol. 39. - P. 901¬906. 68) Williams C.J.,Smith R.A.,Ball R.J.,Wilkinson H. Hypercalcaemia in osteogenesis imperfecta treated with pamidronate. Arch Dis Child. 1997,76:169-70 69) Zacharin M., Bateman J. Pamidronate treatment of osteogenesis imperfecta – lack of correlation between clinical severity, age at onset of treatment, predicted collagen mutation and treatment response. J Pediatr Endocrinol Metab. 2002, 15:163-74 70) Bisphosphonate therapy for children and adolescents with secondary osteoporosis / L.Ward // Cochrane Database Syst. Rev. – 2007, Iss. 4. 71) Bisphosphonate therapy for osteogenesis imperfecta / C.A.Phillipi // Cochrane Database Syst.c Rev. – 2008, Iss. 4.
Информация
Сокращения
, и
спользуемые в протоколе:
| ГФФ | - | Гипофосфатезия |
| НО | - | Несовершенный остеогенез; |
| МПКТ | - | Минеральная плотность костной ткани; |
| ОАК | - | Общий анализ крови; |
| ОАМ | - | Общий анализ мочи; |
| РКИ | - | Рандомизированные клинические исследования; |
| УЗИ | - | ультразвуковое исследование; |
| ЮИО | - | Ювенильный идиопатический остеопороз. |
Список разработчиков протокола:
1) Сатжанов Азат Бекенович - магистр медицины, заместитель главного врача ЖОДБ.
2) Нагыманов Болат Абыкенович - доцент, кандидат медицинских наук, главный внештатный детский травматолог-ортопед МЗ СР РК, заведующий отделением ортопедии №1 ННЦМД.
3) Нурмуханов Ардак Максутович - врач ортопед-травматолог ННЦМД.
4) Сатбаева Эльмира Маратовна - кандидат медицинских наук, РГП на ПХВ "Казахский национальный медицинский университет им. С.Д. Асфендиярова", заведующая кафедрой фармакологии.
Указание на отсутствие конфликта интересов : нет.
Список рецензентов:
1) Нагимтаева Алмагуль Аманжоловна - кандидат медицинских наук, врач - генетик, Филиал Корпоративного фонда «University Medical Center» «Национальный научный центр материнства и детства» г. Астана.
2) Нигматуллина Назым Бахытбековна - кандидат медицинских наук, старший ординадотор отдела нефрологии, диализа и трансплантации Филиал Корпоративного фонда «University Medical Center» «Национальный научный центр материнства и детства» г. Астана.
Прикреплённые файлы
Внимание!
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта", не может и не должна заменять очную консультацию врача. Обязательно обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может назначить нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта" являются исключительно информационно-справочными ресурсами. Информация, размещенная на данном сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший в результате использования данного сайта.